
Open Access, Volume 9
Alpha 1-antitrypsin deficiency in pulmonary langerhans cell histiocytosis: A case report
Antonio Fabozzi1; Dario Angelone1; Giovanna Palumbo2; Giulia de Rose1; Ilaria Cuccaro1; Roberto Romiti1; Gregorino Paone1; Paolo Palange1
1Department of Public Health and Infectious Diseases, Division of Pulmonary Medicine, “Sapienza” University of Rome, Italy.
2Department of Translational and Precision Medicine, Division of Hematology, “Sapienza” University of Rome, Italy.
Antonio Fabozzi
Department of Public Health and Infectious Diseases, Division of Pulmonary Medicine, “Sapienza” University of Rome, Italy
Email: antoniofabozzi40@gmail.com
Received : May 04, 2023,
Accepted : June 27, 2023
Published : June 30, 2023,
Archived : www.jclinmedcasereports.com
Abstract
The association between Pulmonary Langerhans Cell Histiocytosis (PLCH) and Alpha 1-Antitrypsin Deficiency (AATD) is currently undefined; in fact, the largest scientific study available in literature was performed on 50 patients.
A man with a diagnosis of PLCH since 20 years, came to our pulmonary clinic for dyspnea on exertion (mMRC 2). Global spirometry showed mild obstructive ventilatory deficit with air trapping indices on nitrogen washout test. Single-breath DLCO showed a level at the lower limits of normal. HRCT showed panlobular emphysema and multiple pseudo-nodules in continuity with fibrotic striae and traction bronchiolectasis in the upper lobes. Serum alpha1-antitrypsin determination showed intermediate deficiency. Gene sequencing showed heterozygosity for the deficient Z allele (PI*MZ).
Scientific studies of larger case series of patients with PLCH are necessary to define the true prevalence of AATD in this disease and if this association causes more severe functional deficits, because in selected cases treatment with replacement therapy should be considered.
Keywords: Lung Diseases; Alpha 1-antitrypsin; Alpha 1-antitrypsin deficiency; Histiocytosis Langerhans-Cell; Pulmonary Langerhans Cell Granulomatosis; Pulmonary Emphysemas; Panlobular Emphysema.
Abbreviations: Pulmonary Langerhans Cell Histiocytosis; AATD: Alpha-1 Antitrypsin Deficiency; Spo2: Peripheral Oxygen Saturation; DLCO: Diffusing Capacity of the Lungs for Carbon Monoxide; FEV1: Forced Expiratory Volume in the First Second; FVC: Forced Vital Capacity; RV: Residual Volume; CPR: C-Reactive Protein; A1AT: Alpha-1 Antitrypsin.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Fabozzi A (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Fabozzi A, Angelone D, Palumbo G, de Rose G, Ilaria Cuccaro I, Romiti R, Paone G, et al. Alpha 1-antitrypsin deficiency in pulmonary langerhans cell histiocytosis: A case report. Open J Clin Med Case Rep. 2023; 2063.
Introduction
Pulmonary Langerhans Cell Histiocytosis (PLCH) is a rare cystic-nodular interstitial lung disease predominantly affecting the upper lung lobes in young male smokers. The disease presents as a hyperproliferation of Langerhans-like cells in inflammatory clusters with the presence of also eosinophils, neutrophils, and lymphocytes at the level of bronchovascular structures to extension into the lung parenchyma (stellate lesions). It is typically associated with obstructive ventilatory deficit and decreased DLCO.
Alpha1- Antitrypsin Deficiency (AATD) is a genetic disorder characterized, at the level of the lung parenchyma, by a deficient level of alpha1- antitrypsin and subsequent neutrophil elastase hyperfunction (serine proteinases) with damage to connective elastin and destruction of interalveolar septa until the formation of diffuse emphysematous lesions and an obstructive-type ventilatory deficit.
Currently, data on the true prevalence of AATD in patients with PLCH are insufficient to exclude a possible association between the two diseases, because the largest scientific study currently available is on a cohort of 50 patients [1]. In this study, there was no evidence of a significantly higher prevalence of AATD in the population with PLCH compared to the general population. The most rational physiopathological hypothesis for the correlation between the two diseases is the hyperactivity of serine proteinases triggered by A1AT deficiency, particularly neutrophil elastase, which also appear to play a role in the pathogenesis of PLCH [2].
Case Presentation
A 56-year-old Caucasian man, former smoker (23 pack years), with a diagnosis of Langerhans Cell Histiocytosis on lung biopsy since about 20 years and in treatment at the Histiocytosis Center of the Policlinico Umberto I, was referred to the Rare Lung Disease Clinic of the Policlinico Umberto I for a clinical picture characterized by dyspnea for mild exertion (MMRC 2). On chest auscultation, reduced physiological respiratory noises were present bilaterally over the entire lung area, but pathological noises were absent. The cardiological objective examination was normal. SpO2 in room air was 97%. Global spirometry (Figure 1) showed mild obstructive ventilatory deficit (FEV1/FVC 62.8, FEV1 3.07 L - 85%), with air trapping indices at nitrogen wash out test (RV: 154%; RV/TLC: 41.3). DLCO was 23.32 ml/min/mmHg, at the lower limits of normal (77% of predicted). An HRCT was performed (Figure 2) showing centrolobular and paraseptal emphysematous lesions with associated destruction of interlobular and interlobular septa, with greater distribution in the upper lobes and middle lobe. In the upper lobes multiple pseudo- nodular lesions were present in continuity with retracting fibrotic striae and traction bronchiolectasis. Because of the presence of a significant amount of emphysematous lesions, although in nontypical lung regions, we decided to assay serum alpha1-antitrypsin, which was found to be 78 mg/dL (PCR 0.13 mg/dL). These values revealed the presence of an intermediate A1AT deficiency.
AATD Gene sequencing of the SERPIN1 gene was then performed, and heterozygosity for the Z-deficient allele (PI*MZ), p.Glu366Lys mutation (c.1096G>A), was revealed.
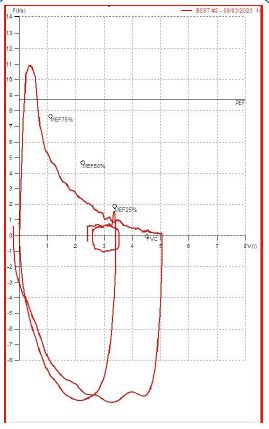
Figure 1: This forced flow-volume curve, correctly performed, showed the characteristic upward concave shape of the expiratory portion of the curve (obstructive ventilatory deficit).
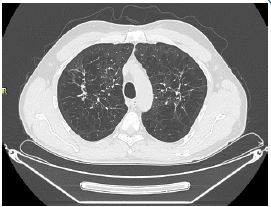
Figure 2: The following HRCT showed the presence in the posterior and anterior segments of the right and left upper lobe of panlobular and paraseptal emphysematous bullae, associated with pseudo-nodular areas in continuity with retracting fibrotic striae and traction bronchiolectasis.
Discussion
It is necessary to perform scientific studies on larger case series of patients with pulmonary Langerhans cell histiocytosis in order to define the true association between PLCH and AATD and its impact on pulmonary function in these subjects. The serum alpha 1-antitrypsin assay and the investigation of a possible mutation in the SERPIN1 gene could become a routine test for patients with progressive and severe PLCH, especially if emphysematous lesions are predominant at HRCT. Thus, treatment with enzyme replacement therapy could be considered in selected cases.
Declarations
Financial/Nonfinancial disclosures: The authors have no conflict of interest to declare in relation to the report.
Acknowledgments: A Fabozzi, G Palumbo, D Angelone, G Paone, P Palange participated to the writing. I Cuccaro, G de Rose, R Romiti participated to the patients’ clinical management.
References
- McCarthy C, Bugnet E, Benattia A, Keane MP, Vedie B, et al. Clarifying the relationship between pulmonary langerhans cell histiocytosis and Alpha 1 antitrypsin deficiency. Orphanet J Rare Dis. 2021; 9; 16: 72.
- Hayashi T, Rush WL, Travis WD, Liotta LA, Stetler-Stevenson WG, et al. Immunohistochemical study of matrix metalloproteinases and their tissue inhibitors in pulmonary Langerhans’ cell granulomatosis. Arch Pathol Lab Med. 1997; 121: 930-937.
