
Open Access, Volume 10
Infantile arterial calcification caused by a novel genetic variant: A case report and literature review
Rima S Bader1; Nedaa Mohammed Bahkali2; Hanin Mohammed Yaqoob Ahmad3*
1Professor of Pediatric and Fetal Cardiology, King Abdulaziz University, Jeddah, Saui Arabia.
2Obstetrics and Gynecology, Assistant professor in King Abdulaziz University, Jeddah, Saudi Arabia.
3Medical Intern at King Abdulaziz University, Jeddah, Saudi Arabia.
Hanin Mohammed Yaqoob Ahmad
Medical Intern, King Abdulaziz University, Jeddah, Saudi Arabia.
Email: hanin.m.y.ahmad@gmail.com
Received : November 11, 2024,
Accepted : December 09, 2024
Published : December 16, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Idiopathic Infantile Arterial Calcification (IIAC) is a rare autosomal recessive disorder primarily associated with variants in the ENPP1 gene that encodes ectonucleotide pyrophosphatase/phosphodiesterase 1. Most cases are diagnosed postnatally. Here, we report a case of prenatal diagnosis of IIAC caused by a novel variant in ENPP1.
The mother was a Saudi female in her mid-30s with a history of first-degree consanguineous marriage; this was her second pregnancy. The fetus was diagnosed at 29 weeks-of-gestation with severe cardiovascular anomalies, including diffuse arterial calcifications, stenosis, myocardial ischemia, heart failure, and bilateral pulmonary hypoplasia. Prenatal echocardiography confirmed extensive calcifications in the thoracic and abdominal aorta, pulmonary trunk, and coronary arteries. Genetic testing of fetal cord blood revealed a previously unreported homozygous missense variant, c.808G>A (p.Glu270Lys), in the catalytic domain of ENPP1; this enzyme produces pyrophosphate, a potent inhibitor of calcification in the body. Both parents carried this variant with a heterozygous genotype. The pregnancy culminated in preterm rupture of membranes at 32 weeks, and the patient delivered a stillborn female infant weighing 2.3 kg. Despite this outcome, the patient had a successful subsequent pregnancy, resulting in the delivery of a healthy boy without ENPP1 variant. This case highlights the significance of early genetic screening and prenatal diagnosis of IIAC, particularly in consanguineous marriages.
The identification of an ENPP1 variant prenatally enabled informed decision-making and perinatal management. The successful outcome of a subsequent pregnancy emphasizes the importance of genetic counseling and individualized prenatal care in cases of IIAC.
Keywords: Generalized arterial calcification of infancy; ENPP1 variant; Fetal hydrops; Myocardial ischemia; Case report.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Yaqoob Ahmad HM (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Bader RS, Bahkali NM, Yaqoob Ahmad HM. Infantile arterial calcification caused by a novel genetic variant: A case report and literature review. Open J Clin Med Case Rep. 2024; 2308.
Introduction
Idiopathic Infantile Arterial Calcification (IIAC) is a rare, life-threatening autosomal recessive disorder characterized by the calcification of medium and large arteries. Most cases (85%) are caused by biallelic variants in the ectonucleotide pyrophosphatase/phosphodiesterase 1-encoding gene ENPP1 on chromosome 6q23.2. A few cases involve mutations in the ABCC6 gene on chromosome 16p13.11 [1]. An adult-onset form of the condition is caused by biallelic variants in NT5E gene located on chromosome 6q14.3 (PubMed: 21288095). The first documented case of IIAC was reported by Bryant in 1901 [2].
systematic review conducted in 2015, which included 200 cases, most of which were diagnosed postnatally, revealed that the majority of affected infants die before the age of 6 months [3]. The estimated prevalence of IIAC is approximately 1 in 390,000 live births [4]. Since 2015, >30 additional cases have been reported in the literature. Although the disease typically presents prenatally, symptoms usually manifest between 3 to 4 months-of-age, often due to calcium deposition and fibrotic changes [5-7]. The most common clinical presentations include respiratory distress, cyanosis, and heart failure [8,9].
IIAC has a poor prognosis, with cardiovascular complications being the primary cause of death in affected individuals [10]. Bisphosphonate therapy has been proposed as a treatment option, but there is conflicting evidence regarding its effectiveness in preventing or reversing arterial and cardiac calcifications [11-13].
Herein, we report a case of IIAC caused by a novel homozygous variant in ENPP1, detected prenatally during the patient’s second pregnancy, which led to a stillbirth.
Case Presentation
Patient History
The patient was a Saudi in her mid-30s. She was married to her first-degree cousin, a healthy male in his mid-30s. Her first Pregnancy (P1), 10 years ago, resulted in a healthy baby delivered through spontaneous vaginal delivery at term with vacuum assistance. The patient’s medical and surgical history was unremarkable. The patient has been deidentified in this report. The case report has been prepared according to the CARE guidelines.
Second Pregnancy (2020)
The patient’s second Pregnancy (P2) was complicated by multiple severe fetal anomalies. At 32 weeks of gestation, she delivered a stillborn baby girl.
A transabdominal fetal echocardiogram performed at 27 weeks of gestation revealed an abnormal cardiac axis, ventricular asymmetry with a smaller right ventricle, and severe calcifications of the tricuspid valve, pulmonary valve, and descending aorta (Figure 1). Cardiac function was borderline, with considerable pericardial effusion and a circumferential myocardial performance index within normal limits for the left ventricle. The four-chamber view was abnormal, with atrioventricular and semilunar valve dysfunction likely secondary to progressive calcification of the great vessels (Figure 2). The fetal heart rate was 145 beats per minute in sinus rhythm.
Given the severe and extensive calcifications, a comprehensive genetic evaluation was undertaken. Amniocentesis and cordocentesis were performed for chromosomal analysis, including fluorescence in situ hybridization and karyotyping, as well as clinical whole exome sequencing. The genetic workup revealed a homozygous missense variant, c.808G>A; p.Glu270Lys, in the ENPP1 gene in the affected individual while the parents were found to be heterozygous. This specific variant has not been reported previously in literature as well as in gnomaAD database (https://gnomad.broadinstitute.org). In support of the association of this variant with the IIAC phenotype in our case, in silico analysis using REVEL tool (PMID: 27666373) predicted the variant to be pathogenic.
A follow-up fetal echocardiogram at 29 weeks demonstrated diffuse echodense calcification involving the entire thoracic and abdominal aorta, the pulmonary trunk, its major branches, and lobar, segmental, and subsegmental intrapulmonary branches, as well as the visualized coronary arteries. Additionally, the tricuspid annulus and its leaflets were severely calcified with restricted movement. The mitral annulus was also calcified with thickening of the anterior leaflet. There was echogenic thickening of the apical trabecular part of the right ventricle and its moderator band, indicating ischemic changes secondary to widespread coronary calcifications.
Consequently, the right ventricle showed near-total loss of function, while the left ventricle exhibited impaired diastolic function with preserved systolic function. Doppler flow study revealed limited tricuspid inflow with trivial regurgitation due to near-total right ventricular akinesia, severe mitral regurgitation, absent pulmonary valve flow (functional pulmonary atresia), abnormal umbilical artery flow (absent end-diastolic flow), and reversed A-wave in the ductus venosus. Fetal heart rate and rhythm remained in sinus rhythm at 140 beats per minute.
The fetal Doppler studies and echocardiographic findings indicated severe congestive heart failure with cardiomegaly, dilated atria, and bilateral pulmonary hypoplasia. These features were consistent with advanced IIAC, leading to significant myocardial ischemia and hypoxia, systemic congestion, and hydrops fetalis.
At 32 weeks of gestation, the patient presented with Preterm Premature Rupture of Membranes (PPROM). Given the critical situation and the poor prognosis, the multidisciplinary team counselled the parents and a decision to proceed with a Do Not Resuscitate order was made. The baby was delivered via assisted breech vaginal delivery and was stillborn, weighing 2.3 kg.
Postmortem findings were consistent with the prenatal diagnosis, confirming severe and diffuse arterial calcifications, myocardial ischemia, and pulmonary hypoplasia. The diagnosis of IIAC, attributed to the ENPP1 gene variant, was confirmed.
Third Pregnancy (2023)
Following the adverse outcome of the second pregnancy, the patient conceived again. Given her history, this third Pregnancy (P3) was closely monitored with a focus on early detection of any genetic or anatomical abnormalities.
A normal nuchal translucency scan at 11 weeks measured 1.37 mm. Amniocentesis performed for genetic analysis, including a targeted gene test, showed no abnormalities. Serial growth and anatomy scans at 18+5 weeks and 25+5 weeks were within normal limits, and a fetal echocardiogram at 25 weeks confirmed normal cardiac anatomy and function.
Throughout the pregnancy, the patient was managed by a multidisciplinary team, including maternal-fetal medicine specialists and genetic counselors. The pregnancy progressed uneventfully, and the patient delivered a healthy baby boy weighing 2.6 kg via spontaneous vaginal delivery. Both mother and baby were discharged home in good condition.
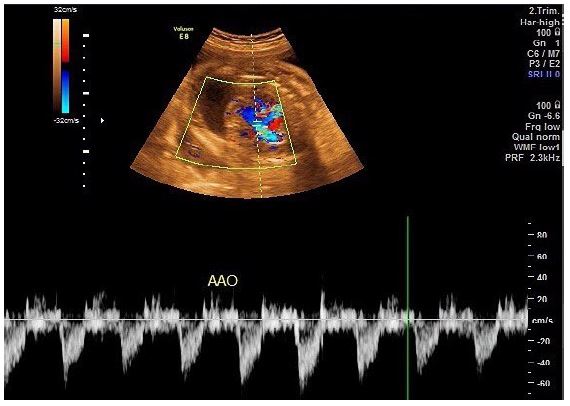
Figure 1: Clinical image.
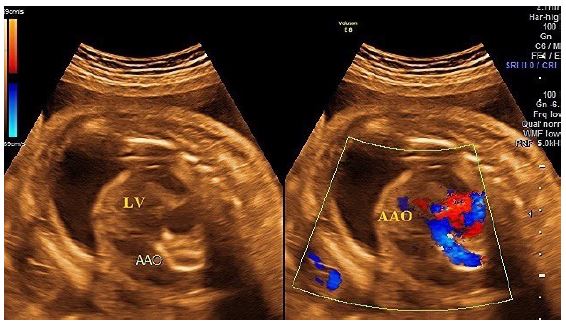
Figure 2: Clinical image.
Discussion
IIAC is a rare autosomal recessive disorder predominantly associated with biallelic mutations in the genes ENPP1 or ABCC6, leading to pathological calcium deposition in arterial walls. This often results in severe cardiovascular complications, including stenosis, myocardial ischemia, and heart failure, and can be fatal in infancy. Most IIAC cases are diagnosed postnatally, with few reported instances of prenatal detection. The current case highlights the genetic underpinnings of IIAC in a consanguineous marriage, emphasizing the importance of genetic testing and early diagnosis.
In this case, the patient, a healthy female with a history of a first-degree consanguineous marriage, experienced a second pregnancy complicated by IIAC, which was detected prenatally at 29 weeks of gestation. The fetus exhibited significant arterial calcifications, stenosis, myocardial ischemia, severe heart failure, and bilateral pulmonary hypoplasia. These findings were consistent with other reported cases of IIAC, where prenatal ultrasound and echocardiography revealed similar severe cardiovascular anomalies.
The role of consanguinity in the inheritance of IIAC is well-documented. A 2023 case report described a similar instance of IIAC in an infant born to parents in a second-degree consanguineous marriage [14]. However, not all IIAC cases are associated with consanguinity, as evidenced by reports of the condition in non-consanguineous families [15,16]. This suggests that while consanguinity increases the risk of IIAC, it is not necessary for the condition to occur.
The findings in this case were comparable to those in a case reported in the USA where echocardiography at 30 weeks of gestation revealed pericardial effusion in a fetus diagnosed with IIAC [17]. A retrospective study involving 10 children diagnosed with generalized idiopathic arterial calcification reported that nearly half of the cases were diagnosed during the prenatal period, highlighting the growing ability to detect IIAC early [18].
Nevertheless, a review by Curtis reported a median age at diagnosis of 3 months [10]. In a case reported by Swati et al., the diagnosis was made postnatally through autopsy [14]. Similarly, a case from Romania confirmed the diagnosis postnatally via histopathological findings and genetic testing [15]. Cases of IIAC present with a range of symptoms, with lower respiratory tract infections, cardiac failure, seizures, hypertension, and hypertrophic cardiomyopathy being the most common postnatal symptoms, and hyperechogenic valves, fetal pericardial effusion, diffuse arterial calcification, stenosis, myocardial ischemia, severe heart failure, and bilateral severe pulmonary hypoplasia being the most common prenatal findings. Fetal hydrops is a common finding and supports the diagnosis of IIAC, with only one reported case of prenatal diagnosis in the absence of fetal hydrops [19].
In the present case, the mother experienced PPROM at 32 weeks, leading to an assisted breech vaginal delivery with a stillbirth weighing 2.3 kg. This was similar to a recent Indian case where vaginal delivery occurred at 30 weeks of gestation with a birth weight of 1.8 kg [14]. Three recent cases were delivered via cesarean section [15,16,20], highlighting the variability in the mode of delivery among mothers with IIAC-affected pregnancies.
Genetic testing in the present case involved cord blood analysis, revealing a karyotype of 46 XX with a normal female chromosomal pattern. A homozygous missense variant of the ENPP1 gene, c.808G>A(p.Glu270Lys), was identified, which has not been reported previously. Both parents carried this variant with a heterozygous genotype. The ENPP1 gene is associated with autosomal recessive disorders including IIAC, autosomal recessive hypophosphatemic rickets type 2, and pseudoxanthoma elasticum. In the literature, there is marked predominance of ENPP1 variants as the cause of IIAC, with a few reported cases involving ABCC6 mutations. For example, a recently reported Chinese case of IIAC involved c.783C>G variant of the ENPP1 gene, a variant that had only been reported in three cases previously [20]. Another case involved c.1112A>T and c.130C>T variants in the ENPP1 gene in a preterm infant. The case was successfully treated with bisphosphonates and antihypertensive agents [21]. In 2018, a 2-month-old Chinese boy presenting with persistent dyspnea, hypertension, proteinuria, and convulsions was diagnosed with IIAC, and was found to have c.130C>T and c.1112A>T variants in the ENPP1 gene [22]. Similarly, Xiaobi et al. reported two Chinese children diagnosed with IIAC, both of whom had variants in ENPP1. The children were treated symptomatically, and while both children were alive at the last reported follow-up, only one responded well to treatment [23].
ENPP1 encodes ectonucleotide pyrophosphatase/phosphodiesterase 1, a 925-amino acid enzyme that cleaves substrates including phosphodiester and pyrophosphate bonds of nucleotides and nucleotide sugars. There is one human transcript of ENPP1 (GenBank accession number NM_006208.3). The main substrate of ENNP1 is extracellular ATP, which is cleaved into AMP and pyrophosphate. Pyrophosphate is a potent inhibitor of calcification in the body [24]; the decrease in pyrophosphate levels as a result of dysfunctional ENPP1 is believed to contribute to the vascular calcification in IIAC [25].
The variant c.808G>A in ENPP1 results in an amino acid substitution from glutamic acid to lysine at residue 270 (p.Glu270Lys), which is in the Phosphodiesterase Catalytic Domain (PCD) of the enzyme. Pathogenic variant of residue 270 has not been reported previously, nor has this variant been studied biochemically. However, inspection of the protein X-ray crystal structure (Protein Data Bank accession code 6WFJ) indicates that residue Glu270 is involved in an extensive hydrogen bonding network that will be disrupted by loss of the carboxylic acid side-chain, which itself forms four hydrogen bonds. Replacement of glutamic acid with lysine also results in a change of the side-chain charge from negative to positive. Therefore, missense variant c.808G>A (p.Glu270Lys) may result in significant structural disruption of the catalytic domain of ENPP1. The consequences of such variants can include decreased protein abundance, decreased enzymatic activity, and impaired cellular localization of the enzyme, all of which can lead to decreased production of pyrophosphate and hence IIAC [25].
Nevertheless, environmental and epigenetic factors are also considered to contribute to disease phenotypes [25]; it remains to be determined via more cases whether that is so for the novel variant reported here.
Treatment strategies for IIAC focus on slowing the progression of arterial calcification, managing cardiovascular symptoms, and improving patient outcomes. The treatment generally involves bisphosphonates, particularly etidronate, due to their ability to inhibit hydroxyapatite crystal formation, a key component in pathological calcification. Studies have shown that etidronate can reduce arterial calcification and improve clinical outcomes, although its long-term efficacy and safety remain subjects of ongoing research [26,27]. In addition to bisphosphonate therapy, the management of IIAC involves symptomatic treatment and addressing the secondary complications associated with the disease, including hypertension and heart failure. Antihypertensive agents, including beta-blockers and angiotensin-converting enzyme inhibitors, may be used to manage blood pressure and reduce the strain on the cardiovascular system [28]. In severe cases, surgical interventions, such as balloon angioplasty or arterial grafting, may be necessary to restore blood flow and prevent organ damage. However, these interventions are associated with significant risks, particularly in the pediatric population, and are generally considered only when pharmacological management has failed [29]. Emerging therapeutic approaches include gene therapy and novel inhibitors targeting the pathways involved in vascular calcification. Advances in genetic testing have enabled early diagnosis and the potential for personalized treatment approaches based on the specific genetic mutations involved in each case [30]. However, the rarity of IIAC poses challenges in conducting large-scale clinical trials, and most current knowledge is derived from case reports and small observational studies.
Conclusions
In this case of a female with a first-degree consanguineous marriage whose second pregnancy was complicated by IIAC, prenatal echocardiography revealed fetal arterial calcifications, stenosis, myocardial ischemia, severe heart failure, and bilateral pulmonary hypoplasia. The pregnancy ended with PPROM at 32 weeks, leading to stillbirth. The case demonstrates the critical role of genetic evaluation in managing pregnancies complicated by severe fetal anomalies, particularly in cases of consanguineous marriage. Here, genetic testing of fetal cord blood identified a novel pathogenic homozygous missense variant in the catalytic domain of ENPP1, an enzyme that produces pyrophosphate, a potent inhibitor of calcification in the body. The specific variant (c.808G>A, p.Glu270Lys) likely disrupts an extensive hydrogen bonding network in the PCD of the enzyme. This variant expands the genetic landscape of IIAC. The case highlights the significance of early genetic screening and prenatal diagnosis of IIAC, to enable informed decision-making and perinatal management, and of genetic counseling, early detection and multidisciplinary care in subsequent pregnancies.
References
- Guimaraes S, Lopes JM, Oliveira JB, et al. Idiopathic infantile arterial calcification: A rare cause of sudden unexpected death in childhood. Path Res Int. 2010; 185314.
- Bryant JH. Case of calcification of the arteries and obliterative endarteritis associated with hydronephrosis in a child aged six months. Guys Hosp Rep. 1901; 55: 17-28.
- Mastrolia SA, Weintraub AY, Baron J, et al. Antenatal diagnosis of idiopathic arterial calcification: a systematic review with a report of two cases. Arch Gynecol Obstet. 2015; 291: 977-986.
- Staretz‐Chacham O, Shukrun R, Barel O, et al. Novel homozygous ENPP1 mutation causes generalized arterial calcifications of infancy, thrombocytopenia, and cardiovascular and central nervous system syndrome. Am J Med Genet A. 2019; 179: 2112-2118.
- Rosenbaum D, Blumhagen J. Sonographic recognition of idiopathic arterial calcification of infancy. Am J Roentgenol. 1986; 146: 249-250.
- Pejovic B, Peco-Antic A, Marinkovic-Eric J. Blood pressure in non-critically ill preterm and full-term neonates. Pediatr Nephrol. 2007; 22: 249-257.
- Ziegler SG, Gahl WA, Ferreira CR. Generalized Arterial Calcification of Infancy. In: GeneReviews. University of Washington, Seattle, Seattle (WA). 2020.
- Akhtar Ali S, Ng C, Votava-Smith J, et al. Bisphosphonate therapy in an infant with generalized arterial calcification with an ABCC6 mutation. Osteoporos Int. 2018; 29: 2575-2579.
- Bolster F, Ali Z, Southall P, et al. Generalized arterial calcification of infancy-findings at post-mortem computed tomography and autopsy. Forensic Sci Int. 2015; 254: e7-e12.
- Chong CR, Hutchins GM. Idiopathic infantile arterial calcification: the spectrum of clinical presentations. Ped Dev Pathol. 2008; 11: 405-415.
- Stuart G, Wren C, Bain H. Idiopathic infantile arterial calcification in two siblings: failure of treatment with diphosphonate. Heart. 1990; 64: 156-159.
- Van Reempts P, Boven K, Spitaels S, et al. Idiopathic arterial calcification of infancy. Calcif Tissue Int. 1991; 48: 1-6.
- Meradji M, De Villeneuve V, Huber J, et al. Idiopathic infantile arterial calcification in siblings: radiologic diagnosis and successful treatment. J Pediatr. 1978; 92: 401-405.
- Mune SB, Gadgil P, Shaikh GB, et al. Idiopathic infantile arterial calcification: An autopsy evaluation. IHJ Cardiovascular Case Rep. 2023; 7: 15-18.
- Fãgãrãşan A, Gozar L, Ghiragosian S-ER, et al. Severe early-onset manifestations of generalized arterial calcification of infancy (mimicking severe coarctation of the aorta) with ABCC6 gene variant—Case report and literature review. Front Cardiovasc Med. 2022; 9: 1032519.
- Gurzu S, Burlacu D, Sánta R, et al. Case Report: Coexistence of generalized arterial calcification of infancy (GACI) and maternal infections with cytomegalovirus and Toxoplasma gondii - unexpected fatal complication in a newborn. Front Pediatr. 2022; 10: 922379.
- Weingarten AJ, Muller A, Langman C, et al. Novel and successful treatment of generalized arterial calcification of infancy in a patient with previously undescribed mutation in ENPP1. Progr Pediatr Cardiol. 2022; 66: 101466.
- Ramirez-Suarez KI, Cohen SA, Barrera CA, et al. Longitudinal assessment of vascular calcification in generalized arterial calcification of infancy. Pediatr Radiol. 2022; 52: 2329-41.
- Yi Y, Tong T, Liu T, et al. Prenatal diagnosis of idiopathic infantile arterial calcification without fetal hydrops. Echocardiography. 2017; 34: 311-314.
- Lu P, Chen J, Chen M, et al. Case report: A rare homozygous variation in the ENPP1 gene, presenting with generalized arterial calcification of infancy in a Chinese infant. Front Cardiovasc Med. 2023; 10: 1105381.
- Yunfeng L, Tongyan H, Jing W, et al. Case report: A novel genetic mutation causes idiopathic infantile arterial calcification in preterm infants. Front Genet. 2021; 12: 763916.
- Liu YF, Han TY, Tong XM, et al. Persistent hypertension for two months in a preterm infant. Zhongguo Dang Dai Er Ke Za Zhi. 2018; 20: 939-943.
- Liang X, Zeng S, Li Y, et al. Analysis of two cases with idiopathic infantile arterial calcification. Zhonghua Er Ke Za Zhi. 2014; 52: 874-6.
- Dedinszki D, Szeri F, Kozak E, et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med. 2017; 9: 1463-1470.
- Ralph D, Levine MA, Richard G, et al. Mutation update: Variants of the ENPP1 gene in pathologic calcification, hypophosphatemic rickets, and cutaneous hypopigmentation with punctate keratoderma. Hum Mutat. 2022; 43: 1183-1200.
- Nitschke Y, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: The face and the mirror. Curr Opin Pediatr. 2012; 24: 470-479.
- Rutsch F, Böyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment for generalized arterial calcification of infancy: first international survey on available treatments. J Am Soc Nephrol. 2011; 6: 573-582.
- Ferreira CR, Xia F, Watson EM, et al. Genetic and clinical delineation of classic pseudoxanthoma elasticum and generalized arterial calcification of infancy: implications for precision medicine. Genet Med. 2018; 20: 388-398.
- Levy RJ, Helfer DM, Cory DA, et al. Balloon angioplasty for critical aortic stenosis in an infant with idiopathic infantile arterial calcification. Am J Cardiol. 1991; 68: 825-826.
- Nitschke Y, Rutsch F. Mechanisms of arterial calcification in patients with pseudoxanthoma elasticum and generalized arterial calcification of infancy. Front Genet. 2017; 8: 14.
