
Open Access, Volume 9
Neuronal intranuclear inclusion disease: Two cases report and literature review
Zhaoyang Yan1; Jing Liang2; Haixia Zhang1; Yuanyuan Liu1; Pengfei Wang1*
1Department of Neurology, Weihai Municipal Hospital of Shandong University, Weihai 264200, China.
2Department of Electrophysiology, Weihai Municipal Hospital of Shandong University, Weihai 264200, China.
Pengfei Wang
Department of Neurology, Weihai Municipal Hospital of Shandong University, 70 Heping Road, Huancui District, Weihai 264200, China.
Tel: +0631-5283979;
Email: wpf5287598@163.com
Received : May 10, 2023,
Accepted : July 04, 2023
Published : July 10, 2023,
Archived : www.jclinmedcasereports.com
Abstract
Neuronal intranuclear inclusion disease is a rare and slowly progressing neurodegenerative disease characterized by extensive eosinophilic endonuclear inclusion bodies in central, peripheral, autonomic nervous system and visceral organ cells. Due to its variable clinical manifestations, it is often misdiagnosed. In recent years, with the development of skin biopsy and molecular genetics, the detection rate of this disease has increased greatly. This article aims to improve the neurologists’ early identification by the clinical manifestations, imaging and pathological features of two patients with sporadic NIID.
Keywords: Neuronal intranuclear inclusion; Diffusion weighted imaging; Electrophysiology; Skin biopsy; NOTCH2NLC.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Wang P (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Yan Z, Liang J, Zhang H, Liu Y, Wang P. Neuronal intranuclear inclusion disease: Two cases report and literature review. Open J Clin Med Case Rep. 2023; 2066.
Introduction
Neuronal Intranuclear Inclusion Disease (NIID) is a rare and slowly progressing neurodegenerative disease characterized by eosinophilic hyaline intranuclear inclusions in the central, peripheral, and autonomic nervous system cells. It has been considered to be a heterogenous disease because of its highly variable clinical manifestations, and its antemortem diagnosis has been difficult. The core triad symptoms in the central nervous system, such as dementia, parkinsonism, and psychiatric symptoms, are proposed as an important clue for the clinical diagnosis of NIID. In addition, it also includes episodic encephalopathy, cerebellar ataxia, muscle weakness, sensory disturbance, and autonomic impairment, including vomiting, bladder dysfunction, syncope, miosis, convulsions, and neuropathy [1]. The specific high-intensity signal in the corticomedullary junction on Diffusion-Weighted Imaging (DWI), the pathological result of skin biopsy combined with genetic testing can contribute to the accurate diagnosis of the disease. Here, we reported two cases of adult NIID patients who have similar imaging and pathological features, although their clinical manifestations are different. Ultimately, they were diagnosed with NIID through genetic testing.
Case Presentation
Case 1: A 70-year-old retired teacher with a high school education presented to the emergency room of our hospital because of abrupt onset of shaking in his right hand and speech disturbance for 8 hours in July 2, 2020. The patient initially presented with involuntary shaking of the right hand and inability to hold the chopsticks, which gradually progressed to unclear articulation and unsteady walking. After 4 hours, the patient experienced nausea and vomiting several times, followed by lethargy, inability to speak. During these attacks, the patient denied seizures, headache, or other neurological defects. The patient had a medical history of "tuberculous pleurisy" over 50 years and had been cured. In 2014, he was admitted to the hospital due to "paroxysmal dizziness". On October 28, 2014, Brian MRI showed dilation of the ventricular system and possible communicating hydrocephalus (Figure 1A-D). After treatment with antiplatelet and lipid-lowering drugs, his dizziness was improved but still occurred intermittently after discharge. Half a year ago, His wife noticed a progressive cognitive decline manifested as recent amnesia but not limited activities of daily living. On May 27, 2020, he underwent "transurethral prostatectomy and cystostomy" in other hospitals due to "benign prostatic hyperplasia". His son and daughter are both in good health. His father died of stomach cancer in his 66, and his mother died of lung cancer in her 72. He has three younger brothers, one of whom is a 67-year-old unmarried elderly person with low intelligence when he was young. The other younger brother with a healthy son is a 65-year-old farmer with decreased memory in the past year and was diagnosed with brain atrophy of unknown etiology. The last brother with a healthy son and daughter is 62-years-old sailor and healthy at present.
Neurologic examination demonstrated lethargy, comprehensive cognitive decline including memory impairment, execution dysfunction and aphasia. Static and postural tremors of hands and ataxia were noted without obvious muscle weakness. Mini-Mental State Examination was 20 (normal threshold, 24/30), Frontal Assessment Battery score was 12 (normal threshold, 16/18). The results of routine laboratory tests were within normal limits. Urinary tests showed increased white blood cell counts and bacterial counts. Brain MRI showed multiple patchy high-intensity signals in bilateral frontal lobes and left temporal occipital lobe on DWI, while equal signals on apparent diffusion coefficient (ADC). Compared with the previous imaging, the lesion area was significantly enlarged, and DWI high signal were distributed along the corticomedullary junction, similar to the “cockscomb pattern” (Figure 1E-H). Urinary ultrasound showed cystitis. Electroencephalogram showed no epileptic discharge. Nerve conduction study (NCS) showed that latency of F-wave of the left tibial nerve was slightly prolonged. Skin sympathetic response (SSR) showed amplitude decreased in the four extremities and latency prolonged slightly in both lower limbs. A histopathological examination of skin biopsy specimens showed eosinophilic hyaline intranuclear inclusions in the fibroblasts and sweat gland cells of the skin tissue (Figures 2A,2B). He did not test for NOTCH2NLC gene. However, Fragile X associated syndrome (FXTAX) was ruled out since the genetic testing for the fragile X chromosome mental retardation gene 1 (FMR1) revealed no abnormal GGC repeat expansion (Figure 2C). 8 months later, the patient was readmitted with «lung infection, urinary tract infection». During this period, he continued to experience intermittent dizziness and unsteady walking.
Case 2: A 63-year-old retired farmer with a primary school education presented to our hospital because of episodic dizziness and mental disorders for 5 years in June 16, 2022. On May 1, 2017, the patient initially presented with paroxysmal dizziness, which lasted several to more than 10 minutes each time, accompanied by a sense of rotation. After the last attack, he experienced mental abnormalities, cognitive impairment, lethargy, occasional nonsense. During these attacks, the patient denied epilepsy, muscle weakness. Brian MRI showed multiple strip like high-intensity signals in bilateral frontal parietal lobe, corpus callosum, and near the posterior horn of lateral ventricle on DWI, while equal signals on ADC. The ventricular system was enlarged and the sulci fissure widened, which was considered as reversible posterior leukoencephalopathy syndrome (Figures 3A-D). After treatment with antiplatelet and lipid-lowering drugs, the patient’s symptoms improved after 3 days. On April 1, 2020, the patient experienced paroxysmal dizziness again, accompanied by a sense of rotation and unstable walking. The dizziness was not related to posture or activity, and there was no numbness, muscle weakness, or mental abnormality. Brian MRI showed that bilateral subcortical high-intensity signals in the frontal and parietal lobes were enhanced compared to before, and the lesion area was enlarged (Figures 3E-H). The above treatment was given again and the symptoms improved after 1 week. On June 16, 2022, the patient experienced mental and behavioral abnormalities, characterized by walking barefoot, inability to recognize family members, frequent yawning, inability to avoid obstacles, inability to communicate, occasional self talk, accompanied by high fever, no muscle weakness, consciousness disorders, or seizures. He had a medical history of «type 2 diabetes, neurogenic bladder associated with diabetes, hypertension, coronary heart disease». Family history is negative.
Neurologic examination demonstrated lethargy, speech disturbance, cognitive impairment, miosis, bilateral Babinski sign positive. Mini-Mental State Examination was 17, Frontal Assessment Battery score was 10. Blood tests were normal apart from hypokalemia of 3.18 mmol/L ((3.5-5.5), hyperlactatemia of 4.56 mmo/L (0.5-2.2) and a mildly elevated neutrophils count of 7.85 x 109/L (1.8-6.3). Procalcitonin was 0.l ng/ml (0-0.06). Urinary tests showed increased protein levels, white blood cell counts, red blood cell counts and bacterial counts. Urine organic acid analysis, amino acid and acylcarnitine spectrum analysis of genetic metabolic disease were within normal limits. Brain MRI showed that the lesion area was enlarged again compared with the previous imaging (Figures 3I-L). Urinary ultrasound showed that the residual urine volume in the bladder is about 40 ml, and there is no obvious abnormality in both kidneys, ureters, or bladder. Electroencephalogram showed no epileptic discharge. NCS showed that Sensory Nerve Action Potentials (SNAP) were decreased in bilateral median nerve, superficial peroneal nerve and sural nerve. Distal Motor Latency (DML) were prolonged in bilateral median nerve. Motor Nerve Conduction Velocity (MNCV) were decreased in bilateral median nerve, tibial nerve, common peroneal nerve and left ulnar nerve. Compound Motor Action Potential (CMAP) were decreased in bilateral common peroneal nerve. The latency of F-wave was prolonged in bilateral tibial nerve. SSR showed abnormalities in both lower limbs. A histopathological examination of skin biopsy specimens showed eosinophilic hyaline intranuclear inclusions in the sweat gland, adipocytes, and spindle cells of the skin tissue (Figures 4 A,B). Additionally, an expansion of 108 GGC repeated in the 5 ‘UTR of the NOTCH2NLC gene was detected by repeat-primed polymerase chain reaction (Figure 4C), further confirming the diagnosis of NIID. After 6 months, she still experienced intermittent dizziness, but no further episodes of mental abnormalities by telephone follow-up.
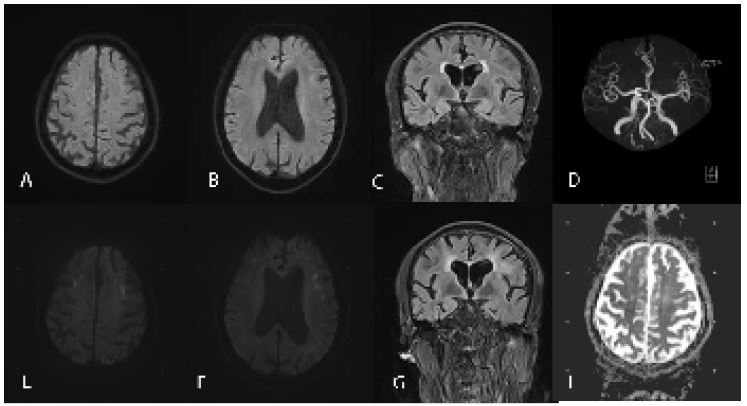
Figure 1: Neuroradiological findings. DWI (A,B), FLAIR (C) and MRA (D) reveals no abnormalities. DWI shows multiple patchy high-intensity signals in the corticomedullary junction of the bilateral frontal lobes and left temporal occipital lobe (E,F). FLAIR shows diffuse high-intensity signal of white matter (G). ADC shows equal signals (H). FLAIR, fluid-attenuated inversion recovery; DWI, diffusion-weighted image; MRA, magnetic resonance angiography.
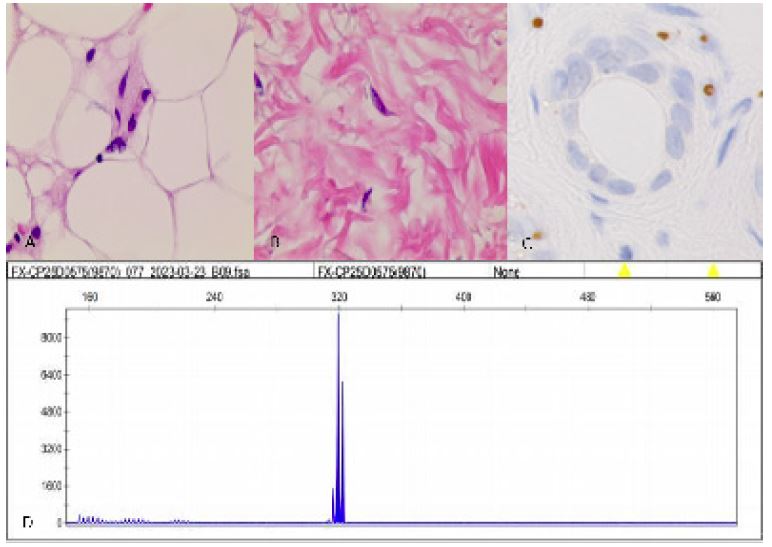
Figure 2: Histopathological and genetic analyses. Hematoxylin-eosin staining shows eosinophilic inclusion bodies in the fibroblasts and sweat gland cells (A,B). The inclusions immunopositive to p62 antibody (C). FMR1 gene revealed no abnormal GGC repeat expansion (D).
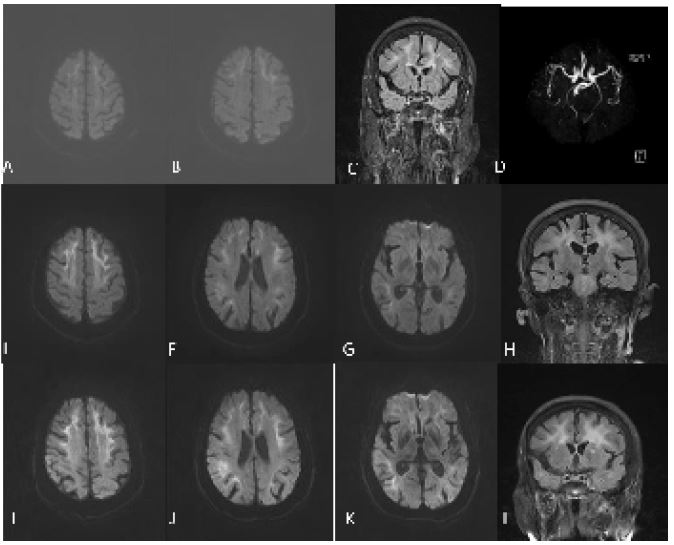
Figure 3: (H) findings. DWI shows multiple strip like high-intensity signals in the corticomedullary junction of the bilateral frontal parietal lobe, corpus callosum, and near the posterior horn of lateral ventricle (A,B). FLAIR shows diffuse high-intensity signal of white matter (C). MRA shows no significant intracranial aortic stenosis or occlusion (D). DWI shows that bilateral subcortical high-intensity signals were enhanced compared to before, and the lesion area was enlarged (E-G). DWI shows the lesion area was enlarged again (I-K).
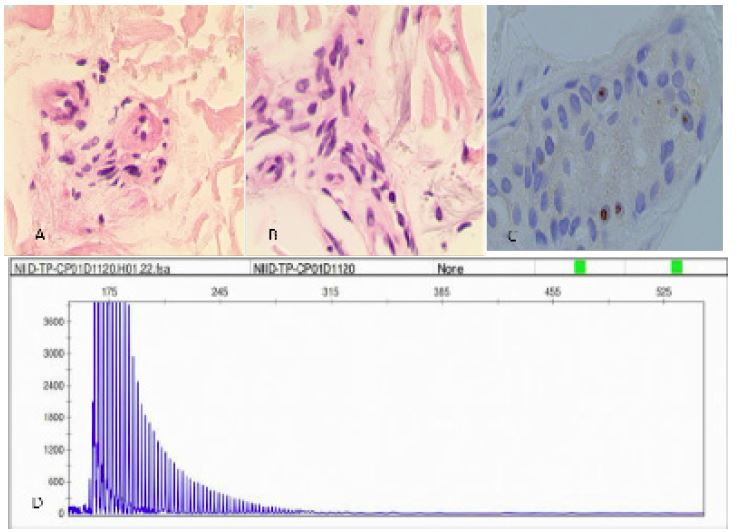
Figure 4: Histopathological and genetic analyses. Hematoxylin-eosin staining shows eosinophilic inclusion bodies in the sweat gland, adipocytes, and spindle cells (A,B). The inclusions immunopositive to p62 antibody (C). RP-PCR of the patient with a characteristic saw-tooth pattern for an expansion of 108 GGC repeated in the 5′ -untranslated region of the NOTCH2NLC Gene (D). RP-PCR, repeat-primed PCR.
Discussion
As early as in 1968, Lindenberg et al. firstly reported a case of a 28-year-old male with clinical symptoms of mental retardation, progressive paralysis, and ataxia, who was identified to have multiple eosinophilic intranuclear inclusions throughout the brain and visceral organs by autopsy. Therefore, they named it “neuronal intranuclear inclusion disease” [2]. NIID is considered to be a slowly progressing neurodegenerative disease but can occur with acute onset. Case 1 showed abrupt onset of speech disturbance and weakness in the right upper limb, which was misdiagnosed as acute cerebral infarction in the early stage. Although DWI revealed multiple patchy hyperintensity lesions in both frontal lobes and left temporal occipital lobe, it does not match the distribution of blood vessels.
Adult-onset patients account for the vast majority of NIID cases in East Asia [3]. So far, only a few juvenile-onset or infantile-onset NIID cases have been described in China [4]. which is vastly different from the reports in European countries. Sone et al. reported that dementia was the most prominent initial symptom in the sporadic NIID cases, followed by miosis, ataxia and unconsciousness. Muscle weakness and sensory disturbance were also observed. While muscle weakness was seen most frequently in familial NIID cases with onset age less than 40 years, followed by sensory disturbance, miosis, bladder dysfunction, and dementia. In familial cases with more than 40 years of onset age, dementia was most prominent [5]. Two cases had different degrees of cognitive impairment, which is consistent with previous report. In case 1, both of the younger brothers had cognitive dysfunction, which could not be ruled out as a familial possibility. Unfortunately, we did not further examine them. Case 2 presented with a subacute progressive encephalitic episode with fever, psychobehavioral abnormality, which is rare in previous case reports [5]. In addition, the patient was accompanied by miosis and neurogenic bladder, which are also common symptoms in patients with NIID and are related to autonomic nervous system damage [6].
Peripheral neuropathy is also reported in patients with NIID. Hong et al. reported that electrophysiological abnormalities were found in 96.4% of NIID patients who presented with central nervous system dominant type but no clinically overt neuropathy, which indicated that subclinical peripheral neuropathy was also common in the specific subgroup of NIID patients. Overall, demyelinating degeneration was the main pathological pattern in these patients, mild axonal degeneration was also observed in some patients [7]. Neuroelectrophysiological examination showed abnormalities in both patients, but there was no evidence of peripheral neuropathy. which is consistent with the above literature reports. Interestingly, both patients showed abnormal SSR in the four extremities, which suggested that SSR could be of great value for early subclinical peripheral neuropathy in NIID patients.
DWI high-intensity signals along the corticomedullary junction became a strong clue for the diagnosis of NIID. The high-intensity signals usually start from the anterior of frontal-parietal lobes; gradually spread to the posterior of frontal-parietal-temporal-occipital lobes with disease advance, and form a “cockscomb pattern” or “ribbon sign” along the corticomedullary junction [8], but did not expand into the deep white matter, even in the advanced stage [5]. However, the imaging feature can also be seen in other nervous system disease. For example, FXTAX has similar clinical symptoms and radiological features to late-onset NIID. They are both autosomal-dominant GGC trinucleotide repeat expansion diseases in different genes (FMR1 gene and NOTCH2NLC gene, respectively) [9]. Therefore, for possible NIID patients with imaging conformity, skin biopsy and genetic testing should be performed early for further confirmation.
NOTCH2NLC is one of the three human-specific NOTCH2-derived genes (NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC) on chromosome 1q21.1 and is highly expressed in a variety of nerve cells, including glia, astrocytes, and microglia. The emergence of NOTCH2NL genes in humans may have contributed to the increase in size and complexity of the human neocortex [10,11]. In 2019, studies reported that repeated GGC expansion in the 5 'UTR of the NOTCH2NLC gene are the major genetic factor leading to the NIID in adults and adolescents [12,13]. Later studies demonstrated that many other neurodegenerative diseases, such as Alzheimer's disease, essential tremor, Parkinson's disease, etc were associated with GGC repeat expansion in the NOTCH2NLC gene [14,15]. On the one hand, these diseases showed similar symptoms as NIID, indicating the symptom heterogenicity of NIID; on the other hand, similar to different variants of the same gene associated with distinct genetic diseases, different lengths of GGC repeat expansion in the NOTCH2NLC gene may cause different diseases with variable phenotypes [12,15]. Therefore, Tian et al. suggested defining a term NIID-related disorders (NIIDRD), which will include NIID and other related neurodegenerative diseases caused by the expanded GGC repeat within human-specific NOTCH2NLC [15].
The two patients presented different onset patterns and clinical symptoms. Case 1 presented with stroke-like symptoms. Case 2 presented with psychiatric abnormalities, cognitive impairment and hyperlactemia, which were early misdiagnosed as hereditary metabolic diseases. However, there is a lack of symptoms such as seizures, liver enlargement, and changes in skin and hair. Moreover, there was no abnormality in urine organic acid analysis and amino acid and acylcarnitine spectrum analysis of genetic metabolic disease. To further elucidate the case from the perspective of monism, we assume a likely diagnosis of NIID which was eventually confirmed by the skin biopsy and genetic analysis.
Conclusion
In summary, NIID should be considered in patients with unexplained cognitive impairment accompanied by tremors, miosis, subclinical peripheral neuropathy, and autonomic dysfunction due to its highly variable clinical manifestations. Especially when typical imaging features are found, the disease should be suspected even if the impairment of cognitive function is not obvious. Further pathological and genetic testing should be performed to confirm the diagnosis.
Declarations
Acknowledgments: The authors have no acknowledgments to report.
Conflict of interest: The authors have no conflict of interest to report.
Data availability: Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.
References
- Sone J, Sobue G. [Neuronal Intranuclear Inclusion Disease]. Brain Nerve. 2017; 69: 5-16.
- Lindenberg R, Rubinstein LJ, Herman MM, Haydon GB. A light and electron microscopy study of an unusual widespread nuclear inclusion body disease. A possible residuum of an old herpesvirus infection. Acta Neuropathol. 1968; 10: 54-73.
- Liang H, Wang B, Li Q, Deng J, Wang L, et al. Clinical and pathological features in adult-onset NIID patients with cortical enhancement. J Neurol. 2020; 267: 3187-3198.
- Xiao F, Tian X, Wang XF. Cerebral Atrophy and Leukoencephalopathy in a Young Man Presenting With Encephalitic Episodes. JAMA Neurol. 2018; 75: 1563-1564.
- Sone J, Mori K, Inagaki T, Katsumata R, Takagi S, et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain. 2016; 139: 3170-3186.
- Chen H, Lu L, Wang B, Cui G, Wang X, et al. Re-defining the clinicopathological spectrum of neuronal intranuclear inclusion disease. Ann Clin Transl Neurol. 2020; 7: 1930-1941.
- Hong D, Wang H, Zhu M, Peng Y, Huang P, et al. Subclinical peripheral neuropathy is common in neuronal intranuclear inclusion disease with dominant encephalopathy. Eur J Neurol. 2023; 30: 527-537.
- Wang R, Nie X, Xu S, Zhang M, Dong Z, et al. Interrelated Pathogenesis? Neuronal Intranuclear Inclusion Disease Combining With Hemiplegic Migraine. Headache. 2020; 60; 382-395.
- Padilha IG, Nunes RH, Scortegagna FA, Pedroso JL, Marussi VH, et al. MR Imaging Features of Adult-Onset Neuronal Intranuclear Inclusion Disease May Be Indistinguishable from Fragile X-Associated Tremor/Ataxia Syndrome. AJNR Am J Neuroradiol. 2018; 39: E100-E101.
- Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell. 2018; 173, 1356-1369.
- Suzuki IK, Gacquer D, Van Heurck R, Kumar D, Wojno M, et al. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell. 2018; 173, 1370-1384.
- Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019; 51: 1215-1221.
- Deng J, Gu M, Miao Y, Yao S, Zhu M, et al. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet. 2019; 56: 758-764.
- Sun QY, Xu Q, Tian Y, Hu ZM, Qin LX, et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain. 2020; 143: 222-233.
- Tian Y, Wang JL, Huang W, Zeng S, Jiao B, et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am J Hum Genet. 2019; 105: 166-176.
- Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large Bcell lymphoma. Blood. 2009; 113: 6069-6076.
- Toumishey E, Prasad A, Dueck G, Chua N, Finch D, et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer. 2015; 121: 716-723.
