
Open Access, Volume 9
The first clinical report of Kleefstra syndrome in an omani patient with classical phenotype
Musallam Al-Araimi*; Kamla Al-Salmani; Nishath Hamza; Hiba Al-Mazrooi; Nawal Al-Buloushi
Department of Clinical Genetics & Counseling, National Genetic Centre, Royal Hospital Muscat, Ministry of Health, Muscat, Sultanate of Oman.
Musallam Al-Araimi
Head of Genetic Clinical and Counseling Services Department, National Genetics Centre, Royal Hospital, Ministry of Health, Sultanate of Oman.
Tel: 0096899311052 & 0096824211428; Email: musallam95@yahoo.com
Received : February 28, 2023,
Accepted : April 05, 2023
Published : April 10, 2023,
Archived : www.jclinmedcasereports.com
Abstract
Background: Kleefstra syndrome is a rare genetic disorder encountered in pediatrics. Its main phenotypic features encompass dysmorphic features, including microcephaly, brachycephaly, and midface hypoplasia. There can also be hypotonia, cardiac defects, mental retardation, and developmental delay. Additional features such as genital anomalies, renal anomalies, hearing problems and recurrent infections can also be observed and vary between different patients [1]. The underlying etiology in the majority of reported cases of Kleefstra syndrome is a microdeletion abnormality involving the long arm of chromosome 9 (9q34.3), which leads to disruption of the euchromatin histone methyltransferase 1 (EHMT1) gene.
Case Presentation: This is the first report of a patient with Kleefstra syndrome in Oman. We identified a two years old female patient with developmental delay, hypotonia, microcephaly, distinctive facial features, anal anomalies, glaucoma, and a cardiac defect. Array comparative genomic hybridization analysis determined that there was a large 1,001 kbp (kilo base pair) deletion in the long arm of chromosome 9q subtelomeric region with breakpoints ch9q34.3 (positions 140,018,931-141,020,389).
Conclusion: This is the first clinical report of Kleefstra syndrome in Oman describing novel phenotypes. The findings identified will contribute to our understanding of Kleefstra syndrome and the genetic basis of the disorder.
Keywords: Kleefstra syndrome; 9q subtelomeric deletion syndrome; Development delay; Case reports.
Abbreviations CGH: Comparative genomic hybridization; Del: Deletion; DNA: Deoxyribonucleic acid; EHMT1: Euchromatin histone methyltransferase 1; Kbp: kilo base pair; Mb: Mega bases; OMIM: Online Mendelian Inheritance in Man; q: Long arm.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Al-Araimi M (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Al-Araimi M, Al-Salmani K, Hamza N, Al-Mazrooi H, Al-Buloushi N. The first clinical report of Kleefstra syndrome in an omani patient with classical phenotypes. Open J Clin Med Case Rep. 2023; 2011.
Introduction
Deletion in the long arm of chromosome 9 (9q34.3) is responsible for a rare genetic disorder known as Kleefstra syndrome (OMIM 610253) [2,1]. There are few reports describing this condition, and they are mostly based on patients with mental retardation [3]. Kleefstra syndrome has been characterized by the core phenotype of developmental delay/intellectual disability, hypotonia, and distinct facial features [2,3]. The underlying genetic abnormality for the majority of these patients is microdeletion in the chromosomal 9q region 34.3 or rearrangements and disruption of the euchromatin histone methyltransferase 1 (Eu-HMTase1), which leads to haploinsufficiency of the EHMT1 gene [2,3].
The exact incidence of this syndrome is not yet known; however, with developments in high throughput technologies, detection has become more common [4]. Because of the widespread application of the fluorescence in situ hybridization technique with the increased number of specific probes and Comparative Genomic Hybridization (CGH) array, a high detection rate has been achieved.
The main phenotypes that are commonly seen with this syndrome are developmental delay, intellectual disability, hypotonia, and distinctive facial features [1,3,4]. Other manifestations, such as cardiac defects, microcephaly, recurrent infections, genital/renal anomalies, hearing impairment, epilepsy, psychiatric problems, tracheobronchomalacia, and vesicoureteral reflux, have also been observed in different percentages of patients [1,4].
Clinical Report
We present the case of a two years old female child who was initially referred to the clinical genetic clinic at the age of one year and three months with the main presenting complaints of developmental delay, facial dysmorphism, hypotonia, congenital cardiac defects, and rectoperineal anus. She was born term through spontaneous vaginal delivery to a primiparous mother of 24 years old and father of 26 years old of a non-consanguineous marriage. At an early neonatal age, the patient had an episode of febrile seizures that was managed conservatively without significant consequences.
Clinical Findings
The pregnancy and delivery were uneventful. The patient’s birth weight was 3.5 kg, length was 50 cm, and head circumference was 36 cm. The patient was found to have a cardiac murmur soon after birth, and echocardiography revealed a tiny patent ductus arteriosus with minimal pericardial effusion, which resolved spontaneously. At the age of seven months, she presented to the emergency department with difficulty passing stool. The clinical evaluation revealed an anteriorly displaced anus, which was surgically corrected (anoplasty with limited transposition of the anus). The patient was referred to the clinical genetic department with complaints of developmental delay and facial dysmorphism.
Developmentally, the patient started to sit without support at 1 year of age and she is unable to roll over bed or crawl. She was unable to stand without support. The clinical genetic assessment revealed microcephaly, brachycephaly, midface hypoplasia, coarse facies, frontal bossing, low-set ears, bluish sclera, carp-shaped mouth, hypertelorism, synophrys, macroglossia, everted lower lip, depressed nasal bridge, and anteverted nares (Figure 1).
Other manifestations included rectoperineal anus, which was surgically corrected at 11 months of age, umbilical hernia, and myopia. Magnetic resonance imaging of the brain showed no abnormalities.
Genetic Characterization
The parents were informed of the patient’s condition, and consent for genetic characterization was obtained. A blood sample was collected to be used as a source of lymphocytes and cell culturing for karyotyping. The cells were cultured, harvested, and analyzed using 400–600 bands resolution. For higher resolution assessment, another blood sample was collected from the patient and sent for array CGH. Subsequently, genomic DNA (deoxyribonucleic acid) was obtained from peripheral blood lymphocytes according to the Chelex method [Barber P. Extraction Protocol]. Ultimately, submicroscopic subtelomeric deletions were detected using array CGH.
Genetic Findings
The patient’s karyotype was 46, XX, which is compatible with an apparently normal female from a cytogenetic point of view. Accordingly, CGH was applied, which revealed a 1,001 kbp deletion at the distal part of the long arm of chromosome 9q34.3 (in the patient in the heterozygous state but not in her parents) with 33 genes involved, of which 17 are OMIM genes. This region is related to the 9q34.3 microdeletion syndrome, which is also known as Kleefstra syndrome (Figure 3).
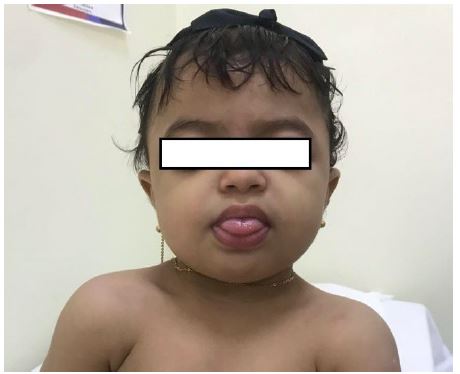
Figure 1: Patient’s clinical features identified: Showing the characteristic facial profile comprising midface hypoplasia, coarse facies, low-set ears, bluish sclera, frontal bossing, carp-shaped mouth, hypertelorism, synophrys / arched eyebrows, macroglossia, everted lower lip, depressed nasal bridge, anteverted nares, and prognathism. Informed consent was obtained for publication of all images present in this manuscript.
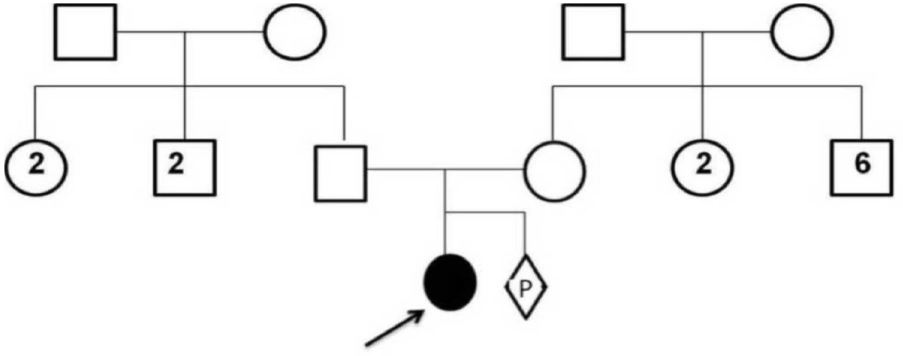
Figure 2: Family pedigree: of the Kleefstra syndrome patient. Three-year-old female patient (indicated by black arrow) and her parents. Proband is an offspring of a non-consanguineous marriage. The numbers shown within squares and circles indicate the number of male and female offspring, respectively.
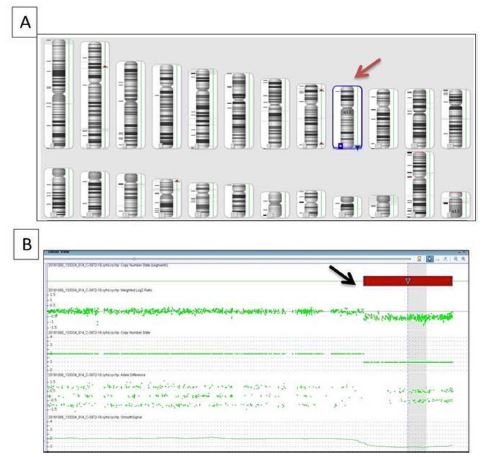
Figure 3: CGH image: Data from array comparative genomic hybridization analysis using ChAS software v3.2. (A) An ideogram where a relative copy number karyotype is used to represent a subtelomeric deletion of 9q (red arrow). (B) SNP array data indicating a 1,001 kbp deletion at the distal part of the long arm of chromosome 9q34.3 (black arrow).
Discussion
Kleefstra syndrome was identified and named by Tjitske Kleefstra in 1993. In 2009, Kleefstra described the distinctive features of the syndrome in more detail. Kleefstra syndrome is associated with a heterozygous deletion of the long arm of chromosome 9 at the 9q34.3 region, which leads to haploinsufficiency of the EHMT1 gene function [2,5].
This phenomenon might occur during translocations by interstitial deletion or in some cases with ring chromosome 9 [6,3,2]. There are 28 exons in the content of the EHMT1 gene, and there is a breakpoint found in intron 9 of the gene with possible deletion / duplication. This impairs protein function, which then presents in patients with Kleefstra syndrome features [5,7,2].
Kleefstra syndrome is a very rare microdeletion syndrome that has been genetically identified and documented worldwide with the development of molecular cytogenetic technologies [8]. The accurate diagnosis of Kleefstra syndrome is essential for patient management, comprehensive care monitoring, and multidisciplinary therapies [8]. Kleefstra syndrome’s clinical features are known to overlap with other genetic syndromes, which makes genotype-phenotype interpretation important in the ultimate diagnosis.
Chromosomal karyotyping analysis was performed at the level of 400-600 bands resolution, and it showed normal female karyotype 46, XX. CGH analysis revealed a 1.211 Mb (mega bases) deletion with the breakpoints of 1,001 kbp deletion at the distal part of the long arm of chromosome 9q34.3.
Yatsenko et al. defined a minimal critical region of about 700 kb deleted in all of the Kleefstra syndrome patients included in their study [9]. The broad range of clinical features identified in different Kleefstra syndrome patients has been attributed to the contiguous gene deletion phenomenon.
Submicroscopic deletion del(9)(q34.3) is a rare constitutional microdeletion syndrome involving a gene-rich region [10]. This region involves deletion of the EHMT1 gene and its haploinsufficiency, which might disturb some of the epigenetic stability signaling pathways of the cell, impairs normal protein function, and plays a key role in Kleefstra syndrome. This is reflected in the genetic features, and representation occurs with the 9q deletion syndrome, known as Kleefstra syndrome [11]. It has been reported that one of the main causes for breakpoints and subsequent deletions / duplication in the EHMT1 gene in Kleefstra syndrome patients is the non-homologous recombination in EHMT1 [7].
Conclusion
This report documents the clinical and molecular findings of a rare microdeletion syndrome in a patient from the Sultanate of Oman. These findings will contribute to our understanding of Kleefstra syndrome and the genetic basis of the disorder. Previous studies have reported patients with phenotypes associated with Kleefstra syndrome in the region [11]. However, this is the first case of Kleefstra syndrome reported in Oman with molecular cytogenetics confirmation, and it has implications for genetic defects of Kleefstra syndrome in the entire region. Identifying and documenting Kleefstra syndrome cases will have a significant impact and is advantageous for the management of families with Kleefstra syndrome patients. Ultimately, it will help to offer these families the option of pre-implantation genetic screening approaches for their next pregnancy.
Declarations
Acknowledgments: The authors acknowledge the support of the National Genetics Centre of the Royal Hospital in Muscat, Oman, without which this case report would not have been completed.
Funding: No funding source to disclose.
Declaration of conflicting interests: None declared. [The authors of this article have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript].
Patient consent: Due permission was obtained from the parents of the patient to publish the case and the accompanying images.
Ethical approval to present this clinical report was obtained from the ethics and research Committee relevant authorities.
References
- Hadzsiev K, Komlosi K, Czako M, Duga B, Szalei R, et al. Kleefstra syndrome in Hungarian patients: additional symptoms besides the classic phenotype. Mol Cytogenet. 2016; 9: 22.
- Willemsen MH, Beunders G, Callaghan M, de Leeuw N, Nillesen WM, et al. Familial Kleefstra syndrome due to maternal somatic mosaicism for interstitial 9q34.3 microdeletions. Clin Genet. 2011; 80: 31-38.
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006; 79: 370-377.
- Kleefstra T, de Leeuw N, Wolf R, Nillesen WM, Schobers G, et al. Phenotypic spectrum of 20 novel patients with molecularly defined supernumerary marker chromosomes 15 and a review of the literature. Am J Med Genet A. 2010; 152A: 2221-2229.
- Rump A, Hildebrand L, Tzschach A, Ullmann R, Schrock E, et al. A mosaic maternal splice donor mutation in the EHMT1 gene leads to aberrant transcripts and to Kleefstra syndrome in the offspring. Eur J Hum Genet. 2013; 21: 887-890.
- Balemans MC, Ansar M, Oudakker AR, van Caam APM, Bakker B, et al. Reduced euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev Biol. 2014; 386: 395-407.
- Schwaibold, EMC, Smogavec M, Hobbiebrunken E, Lorenz Winter L, Zoll B, et al. Intragenic duplication of EHMT1 gene results in Kleefstra syndrome. Mol Cytogenet. 2014; 7: 74.
- Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. Am J Med Genet. 2009; 46: 598-606.
- Yatsenko SA, Cheung SW, Scott DA, Nowaczyk MJ, Tarnopolsky M, et al. Deletion 9q34.3 syndrome: genotype-phenotype correlations and an extended deletion in a patient with features of Opitz C trigonocephaly. J Med Genet. 2005; 42: 328-335.
- Yatsenko SA, Brundage EK, Roney EK, Cheung SW, Chinault AC, et al. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet. 2009; 18: 1924-1936.
- Noruzinia M, Ahmadvand M, Bashti O, Reza Salehi Chaleshtori A. Kleefstra syndrome: the first case report from Iran. Acta Med Iran. 2017; 55: 650-654.
